

界面新闻记者 |
界面新闻编辑 | 谢欣
临床试验,即以人体(患者或健康受试者)为对象的系统性试验,是药物获批上市的必经之路,也是确定药物疗效和安全性的金标准。日前,国家药监局(NMPA)接连发布两份有关药物临床试验机构监督检查、药物临床试验期间安全信息评估与风险管理工作的文件,旨在进一步规范药物临床试验相关工作。
可以预见的是,更科学、完善的药物临床试验监管办法、工作程序将提高临床试验质量,推进药物高水平研发。另外,两份文件都关系到临床试验进度。而在竞争激烈的当下,药物研发进度又直接影响往后的商业化前景。换而言之,这也将使药企在选择临床试验机构和进行试验时更加谨慎,或将增加药企的合规成本。
具体而言,7月3日,国家药监局综合司发布《药物临床试验机构监督检查办法(试行)(征求意见稿)》(下称“《监督检查办法》(征求意见稿)”)。7月10日,国家药监局药品审评中心(CDE)发布《药品审评中心药物临床试验期间安全信息评估与风险管理工作程序(试行)修订稿(征求意见稿)》(下称“《工作程序》(征求意见稿)”)。
在我国,药物临床试验监管方面的一个重要转折出现在2019年。该年,新修订的《药品管理法》正式实施。其中,药物临床试机构实施备案管理、药物临床试验默示许可等制度获得法律地位。
在鼓励新药研发的浪潮下,我国药物临床试验及机构数量猛增。截至目前,全国已有备案的药物临床试验机构1300余家。这一数字在2015年仅有400余家。

另据制药网的数据,2022年,中国1类新药的新药临床试验申请(IND)受理品种为944个品种(共1668件IND注册申请)。其中,1-6月,化药创新药IND申报适应证重点布局在肿瘤、血液疾病、心血管及消化领域;生物创新药IND适应证重点分布于肿瘤,血液疾病及免疫领域。从目前生物药IND分子类型上看,申报重点集中于单抗、双抗、ADC、FC融合蛋白、干细胞疗法及CAR-T疗法。
大幅扩容之下,已有乱象显现。今年6月初,中国临床肿瘤学会副理事长秦叔逵教授在2023抗肿瘤创新药物临床研究论坛上直言,国内大多数医院临床研究水平不够。有的医院一年接400到500项临床试验,但医生不做临床研究,全部交给CRC(临床协调员)、CRA(临床监查员),研究质量堪忧。
对此,本次发布的《监督检查办法》(征求意见稿)即在检查对象上有所侧重。其中第九条指出:
对试验机构、试验专业或者研究者存在以下情形的,应当纳入检查重点:
(一)既往存在不合规问题的;
(二)同期承担临床试验项目较多等可能影响试验质量的;
(三)投诉举报或者其他线索提示存在质量安全风险的。
对此,俄罗斯工程院外籍院士、昆翎医药联合创始人张丹对界面新闻表示,这体现了《监督检查办法》(征求意见稿)提及的,检查内容可以基于风险选择重点。
他指出,前述三种情形无不提示了存在的高风险。例如,对于同期承接临床试验较多的机构,体制内研究人员数量不足以覆盖,势必需要体制外人员协助,这就带来了对后者的管理难题。同时,一旦存在试验流程、设施不规范,经验不足的情况,受到影响的受试者数量众多,因此风险很高,会成为监管部门关注的重点。
此外,张丹还举例了一些特殊情况。例如,有试验机构刚完成备案就承接了大量项目;或承接的是基因治疗等高风险项目;或同类项目在国外出现大量死亡病例等,都可能引起监管部门对试验机构的关注。
在检查结果和处理方式上,检查结果关系到临床试验机构的资质问题,也就是会间接影响的申办方(即药企)的研发进度。
据《监督检查办法》(征求意见稿)第二十五条,根据检查发现缺陷的数量和风险等级,检查综合评定结论将分为符合要求、基本符合要求和不符合要求。
据《监督检查办法》(征求意见稿)第三十四条和三十五条:
对结论为“不符合要求”的试验机构或者试验专业,药品监督管理部门将取消备案。自被
标识 取消备案之日起,试验机构或者试验专业不得开展新的药物临床试验,已开展的药物临床试验不得入组新受试者。对结论为“基本符合要求”的试验机构或者试验专业,药品监督管理部门视情形采取风险警示、告诫、约谈等措施,督促试验机构和研究者持续完善质量管理体系。对存在可能影响受试者安全或者试验数据质量风险的,可以采取限期整改措施,限期整改的时限通常为6个月,相关试验机构或者试验专业不得开展新的药物临床试验。
张丹认为,这一处理措施即在鼓励申办方选择临床合作对象时,就基于该文件主动做风险评估、合规稽查,谨慎选择高标准、低风险的临床试验机构。若发现已经处于试验中的机构不合规,可能的选择有将不合规的机构数据剔除后,看试验是否还能得到强阳性结果,或者增加其他临床实验中心,最坏的结果则是重开临床试验。但张丹提醒,这些补救措施的前提都是首先与监管部门充分沟通,使试验方案获得监管部门的认可。
实际上,前述“基于风险”的监管思路此前已广泛被欧美等国接受。2016年11月,国际人用药品注册技术协调会(ICH)发布药物临床试验质量管理规范ICH-GCP E6(R2),提出建立基于风险的质量管理体系。我国加入ICH后,这一理念也在实际工作中有所涉及。
此前,CDE制定了《药品审评中心药物临床试验期间安全信息评估与风险管理工作程序(试行)》,并于2021年1月在中心内部发布实施。本次发布的《工作程序》(征求意见稿)即是在前者基础上的修订和公开征求意见。
《工作程序》(征求意见稿)明确,药审中心通过临床试验期间安全风险管理系统开展安全信息监测、评估和风险管理工作。可疑且非预期的严重不良反应(SUSAR)报告、研发期间安全性更新报告(DSUR)报告、其他潜在严重安全性信息等均可被导入至风险管理系统中,由临床试验管理处开展规范性审核和风险评估。
对于申办者实施风险管理措施不充分的情形,药审中心将提出进一步的风险控制要求,如风险管理告知、一般风险管理措施、责令暂停临床试验和终止临床试验等。
而据CDE发布的《2021年度药品审评报告》,2021年,即内部实施首年,CDE收到国内临床期间SUSAR首次报告7197份,同比增长54.51%;收到DSUR报告2568份,同比增长42.82%。发出临床试验风险管理告知信86份、临床试验风险控制通知书21份,暂停临床试验通知书1份,建议申办者主动暂停临床试验5次。
可以发现,2021年,CDE暂停临床试验的情况很少,更没有终止临床试验的情况。
对于本次公开的《工作程序》(征求意见稿),张丹认为,这一文件有“未雨绸缪”的意味。原因在于,一方面,国内药物研发正在从仿制转向创新,相较于作用机理、风险获益更清晰、为人们所熟悉的仿制药、Me-too(同类仿创)药,监管和申办方将面对更多细胞基因治疗等新疗法、新靶点的原创新药。另一方面,当下出现更多对于老年人、儿童等特殊群体的临床试验,监管和申办方面对的试验人群挑战也将越来越复杂。因此,药监部门需要先把这一系统建立起来,以应对未来的挑战。
换而言之,《工作程序》(征求意见稿)重点影响的即是创新药企业。
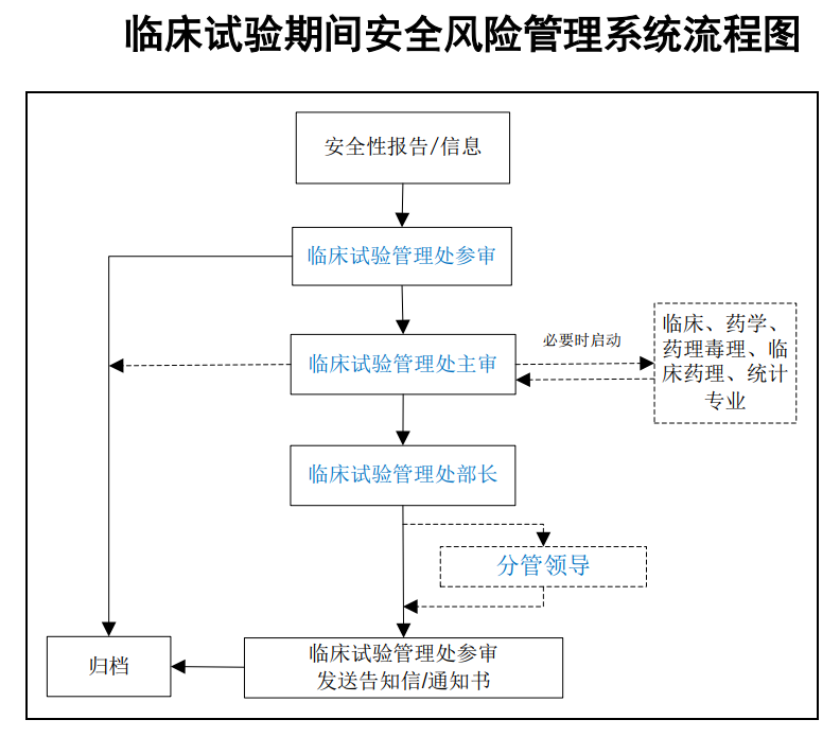
不过,张丹提到,尽管这一文件在CDE收集、判断临床试验过程中的风险信号、各种情况下的处理方法、与申办方的沟通机制等方面都有所阐述,比较详细、可落地执行,从申办方的角度看是“能看明白的”,但具体到各家产品,临床试验风险的阈值和界限是怎样的,还有待进一步明确。
举例来说,急性肝衰竭在人群中的死亡率本身就比较高。针对这一疾病开发药物的临床试验中出现患者死亡,与在皮肤科疾病等低死亡风险临床试验中出现病例死亡相比,显然是不一样的。张丹认为,具体的细化标准未来还可能会以不同的药物开发指南予以澄清。
此外,张丹提到,《工作程序》(征求意见稿)已经很接近美国食药监局(FDA)等国际标准。不过,监管部门在安全信息评估与风险管理的反应时效上还有进一步优化空间。这一方面涉及强制建立电子申报系统。另一方面,若在中国进行的临床试验是国际多中心临床试验的一部分,当海外出现严重不良反应,尤其是死亡病例时,这一满足ICH E2申办条件的执行状况还要多予以关注,即及时获得海外信息,使国内对产品情况有综合的判断。
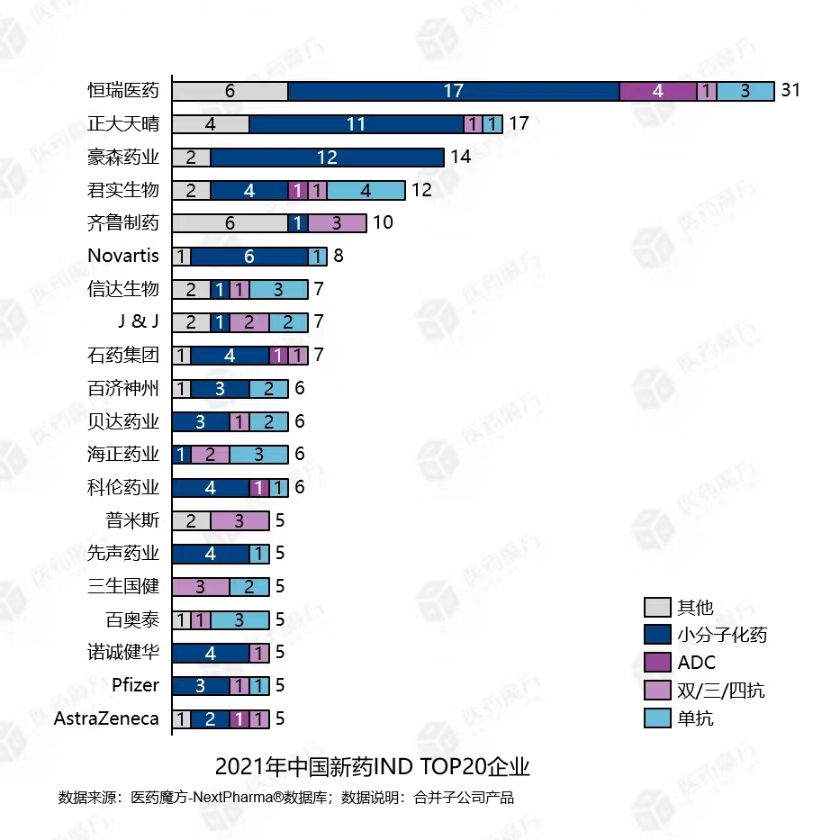
就当下的管线布局而言,医药魔方数据显示,2021年,恒瑞、正大天晴、豪森、君实、齐鲁等头部企业新药申报数量居前。
评论