

记者 |
编辑 | 许悦
3月23日,美国食品药品监督管理局(FDA)官网数据库显示,FDA已授予吉利德科学在研抗病毒药物瑞德西韦(remdesivir)孤儿药资格(Orphan Drug Designation),所涉及的适应症为新型冠状病毒肺炎(COVID-19)。
一般来说,FDA的孤儿药认定主要针对罕见病,FDA对于罕见病的定义是在美国患病人数小于20万人的疾病,由于罕见病药物开发难度大,人群/市场小,药企难以收回成本,开发动力小,孤儿药资格认定及其一系列鼓励措施本质上是为了鼓励针对罕见病的研发。
实际上,早在2015年FDA就已给予瑞德西韦在治疗埃博拉病毒这个适应症上的孤儿药认定,但最终由于瑞德西韦治疗埃博拉病毒的三期临床试验失败,未能获批上市。此次吉利德则在新型冠状病毒适应症上再次提出了孤儿药认定的申请,并获得认定。
对于药物开发者来说,获得FDA孤儿药认定可获得从药物上市到上市后的一系列优惠。包括:
- 税收抵免:孤儿药临床试验费用的50%可以作为税收抵免,并可向前延伸3年,向后延伸15年。
- 免除新药申请费用:FDA认定的孤儿药的新药申请(NDA)费用可以免除。
- 研发资助:FDA从1983年起通过孤儿药资助计划对相关临床研究进行资助。Ⅰ期、Ⅱ期、Ⅲ期临床研究可以获得数十万美元/年的补助。
- 加速审批:开辟孤儿药审批“绿色通道”,对获得孤儿药认定的药物进行加速审批。
- 市场独占权:已认定的孤儿药经FDA批准上市后可享有7年的市场独占权,时间从获批上市为起始时间开始计算,该保护不受专利的影响。在此期间,FDA不再批准针对相同适应证的同分子结构药物,除非原先获批药品被撤回,或该药的持有者许可,或该药供应不足,或新申报的药物更具临床优越性。
- 此外还包括减免处方药用户费用,获得优先审评券等优惠政策。

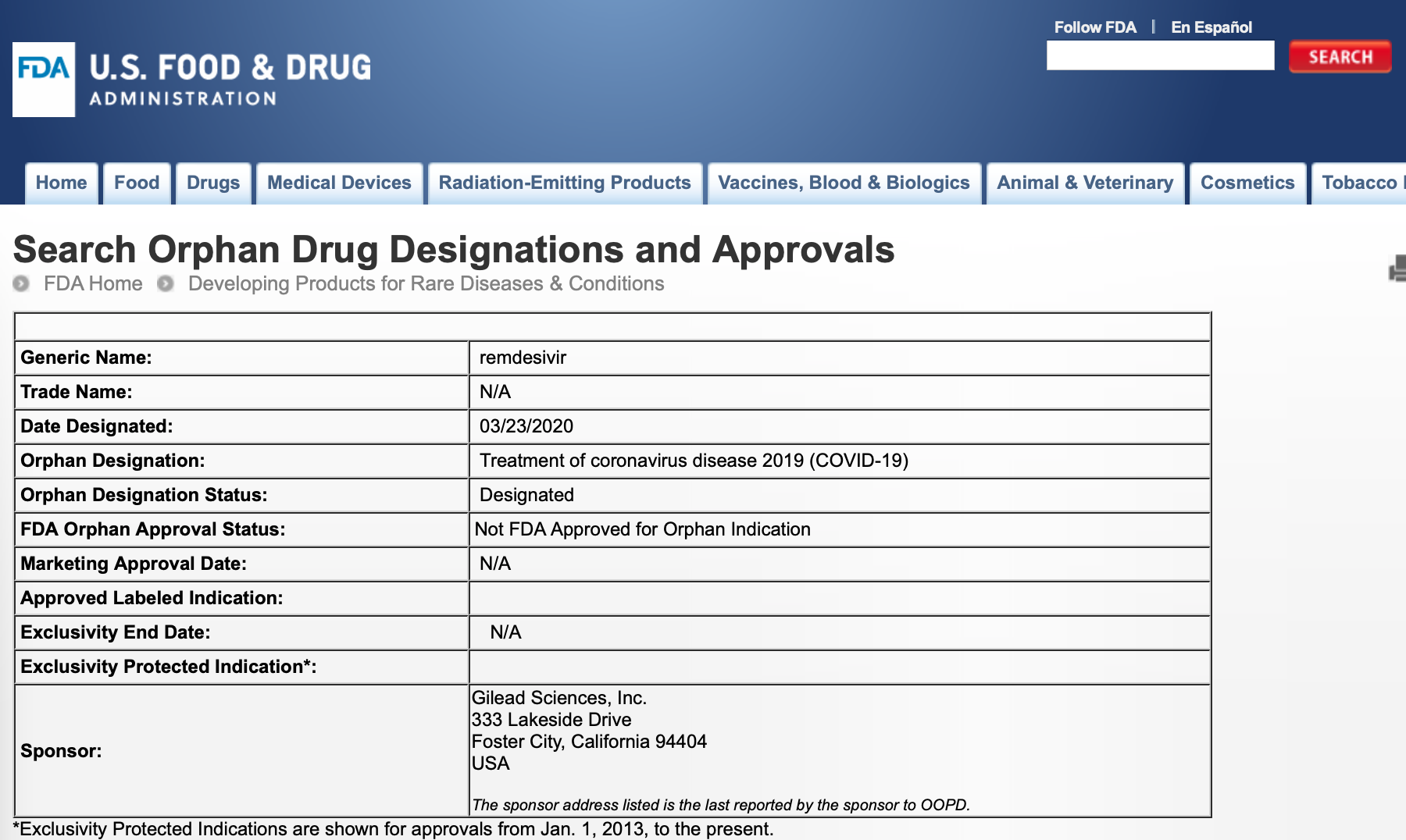
FDA对于孤儿药的注册采取“先孤儿药认定,后上市审批”的方式,目前瑞德西韦依然是一款未上市药品,其对新冠病毒肺炎的疗效仍处于临床试验阶段。
一位医药政策方面专家对界面新闻记者指出,FDA孤儿药认定一般不用于传染病,目前看来,孤儿药资格认定可能更多是商业选择。而未来一旦瑞德西韦疗效被临床试验所证明,FDA通过“紧急授权”的形式(Emergency use authorizations,EUA)批准瑞德西韦上市将是更快的方式。
据介绍,根据美国相关法律规定,类似此次新冠疫情(已被卫生部认定为重大公共卫生事件),FDA可通过紧急授权的形式允许医疗产品(Medical countermeasures,MCM)使用,无论此前该产品是否被批准或者其适应证是否被批准。相比其拓展使用(Expanded Access),即大众所知的同情用药,FDA批准的EUA的实例并不多。据统计从2005至今批准的EUA仅有60多件。
而在新冠疫情暴发后,FDA已经通过EUA形式批准了罗氏诊断与赛默飞世尔的病毒检测产品上市。
除了EUA途径外,根据吉利德3月23日声明,目前全球已有数百位新冠病毒确诊患者接受了瑞德西韦同情用药,这也意味着,使用真实世界证据也可能是未来瑞德西韦加速上市的一种方式。
而吉利德在3月23日的声明中也提及,吉利德目前正在从个人的同情用药请求过渡到扩展使用项目。这种方法既可以加快重症患者获得使用瑞德西韦的速度,又可以收集所有参与患者的数据。不过并不确定这些收集的数据是否有被用于真实世界研究的打算。
从加速上市的角度考虑,中日友好医院曹彬教授在武汉金银潭医院领导开展的瑞德西韦临床试验,作为全球最先开始,也会是最早结束的瑞德西韦治疗新冠病毒肺炎的临床试验,也有存在被纳入全球多中心临床试验的可能性。
评论